
How SARS-CoV-2 immune responses vary by population due to environmental and genetic factors
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2)-infected individuals display a wide range of clinical variations, from asymptomatic infection to lethal disease. In a recent study published on the bioRxiv* preprint server, and international team of researchers investigated the genetic, immunological, and evolutionary factors that determine the vast variability observed in clinical manifestations of the coronavirus disease 2019 (COVID-19).

Study: Environmental and genetic drivers of population differences in SARS-CoV-2 immune responses. Image Credit: Billion Photos / Shutterstock.com
Background
Numerous epidemiological and genetic studies have elucidated the impact of genetic factors and variations in innate immunity, including inborn errors or neutralizing auto-antibodies against type I interferons (IFNs) to contribute to the various SARS-CoV-2-related clinical manifestations. The importance of ancestry-related differences in transcriptional responses to immune challenges have also been described.
Combined with evidence suggesting that viruses and other infectious agents have had an overwhelming impact on human evolution, there remains an urgent need for in-depth investigations of the magnitude of variation in immune responses to SARS-CoV-2 and its drivers across populations worldwide.
For example, in East Asians, strong genetic adaptations starting about 25,000 years ago have been reported against multiple human coronavirus-interacting proteins. Furthermore, many examples of human adaptation to ribonucleic acid (RNA) viruses as a source of population genetic differentiation have also been published.
There is also growing evidence of COVID-19 severity modulations in modern Eurasians due to Neanderthal haplotypes, which are key determinants of their introgression and immunity. Together, this data has sparked curiosity in the scientific community about how these past natural selection events and archaic admixtures might influence the immune response to SARS-CoV-2 in contemporary humans.
About the study
In the present study, researchers used single-cell RNA sequencing (scRNA-seq) with population genetics approaches to characterize cell type-specific transcriptional responses in peripheral blood mononuclear cells (PBMCs) of 222 healthy donors of various ancestries stimulated by SARS-CoV-2 or influenza A virus. PBMCs treated for six hours exhibited strong immune responses and high cell viability, which, in turn, helped the team capture over one million high-quality single-cell transcriptomes.
The study population comprised 80, 80, and 115 individuals of Central African, West European, and East Asian descent, respectively, representing different genetic ancestries owing to their exposure to varying environmental conditions. The effects of human genetic variants on transcriptional variations were assessed by mapping expression quantitative trait loci (eQTLs) that focused on cis-regulatory variants.
The researchers also explored the contribution of natural selection to population differentiation of immune responses. To this end, they searched for overlaps between eQTLs or reQTLs and genome-wide signals of local adaptation that were measured by the population branch statistic (PBS).
In addition, the contribution of differences in cellular proportions to the observed interindividual variability of SARS-CoV-2 responses was determined by focusing on individuals of Central African and West European ancestries, all of whom were recruited during the same sampling campaign.
Finally, the functional consequences of Neanderthal introgression on present-day immune responses to viral challenges was studied. A set of 100,345 introgressed ‘archaic’ alleles or archaic single nucleotide polymorphisms (aSNPs) were used to determine whether eQTLs were over or underrepresented among introgressed variants relative to randomly matched SNPs. Taken together, this information allowed the researchers to understand the contributions of genetic variants altering responses to SARS-CoV-2 in vitro to COVID-19 risk in vivo.
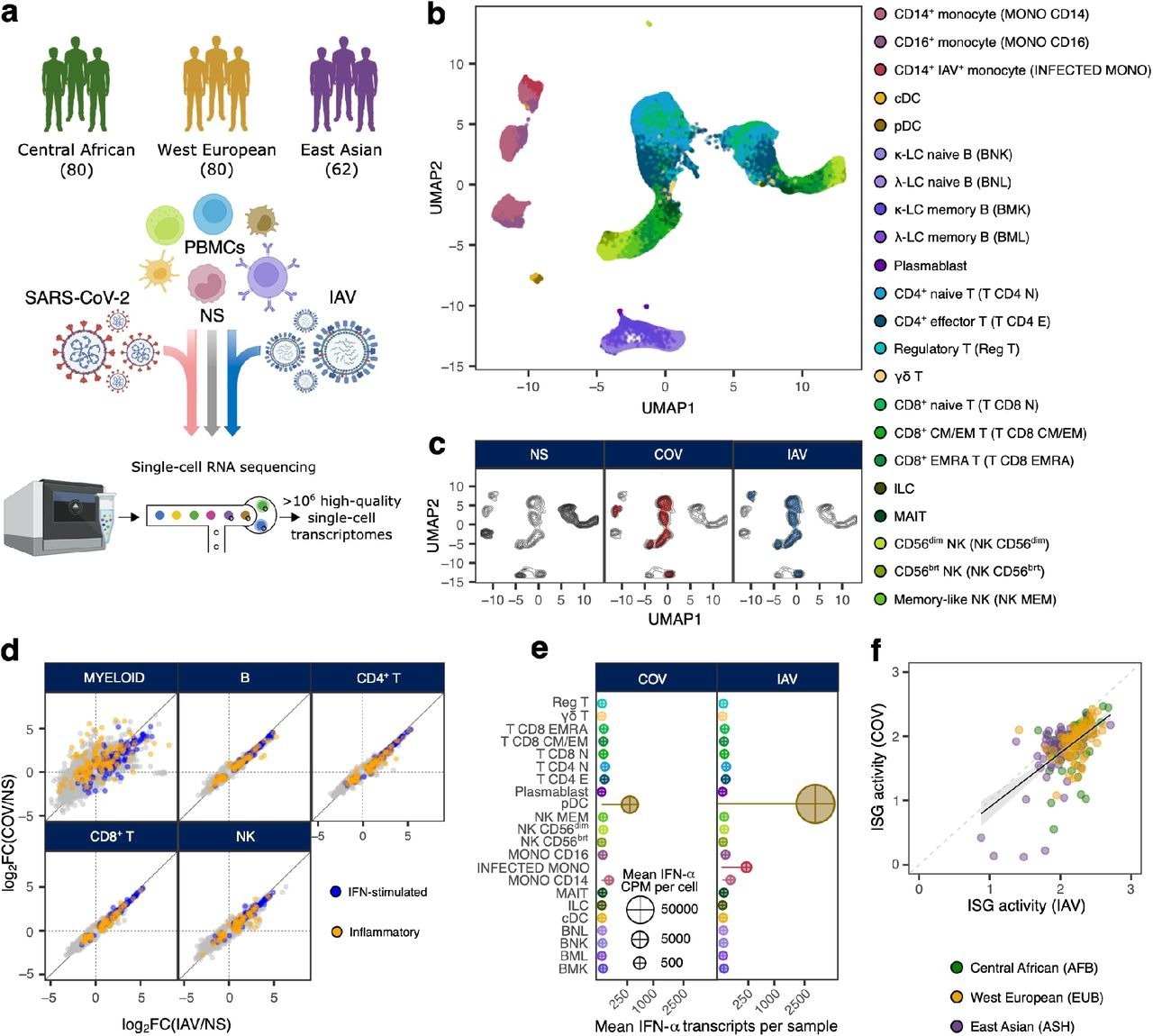 Population-scale single-cell responses to SARS-CoV-2 and IAV. a, Study design. b and c, Uniform manifold approximation and projection (UMAP) of 1,047,824 peripheral blood mononuclear cells: resting (non-stimulated; NS), stimulated with SARS-CoV-2 (COV), or influenza A virus (IAV) for six hours. b, The colors indicate the 22 different cell types inferred. c, Distribution of cells in NS, COV and IAV conditions on UMAP coordinates. Contour plot indicates the overall density of cells, and colored areas delineate regions of high cell density in each condition (gray: NS, red: COV, blue: IAV). d, Comparison of transcriptional responses to SARS-CoV-2 and IAV across major immune lineages. Hallmark inflammatory and interferon-stimulated genes are highlighted in orange and blue, respectively. e, Relative expression of IFN-α-encoding transcripts by each immune cell type in response to SARS-CoV-2 and IAV. Bar lengths indicate the mean number of IFN-α transcripts contributed by each cell type to the overall pool (cell type frequency × mean number of IFN-α transcripts per cell). Dot area is proportional to the mean level of IFN-α transcripts in each cell type (counts per million). f, Correlation of ISG activity scores between individuals, following exposure to SARS-CoV-2 and IAV. Each dot corresponds to a single individual (n = 222) and its color indicates the self-reported ancestry of the individual concerned (AFB: Central African; EUB: West European; ASH: East Asian).
Population-scale single-cell responses to SARS-CoV-2 and IAV. a, Study design. b and c, Uniform manifold approximation and projection (UMAP) of 1,047,824 peripheral blood mononuclear cells: resting (non-stimulated; NS), stimulated with SARS-CoV-2 (COV), or influenza A virus (IAV) for six hours. b, The colors indicate the 22 different cell types inferred. c, Distribution of cells in NS, COV and IAV conditions on UMAP coordinates. Contour plot indicates the overall density of cells, and colored areas delineate regions of high cell density in each condition (gray: NS, red: COV, blue: IAV). d, Comparison of transcriptional responses to SARS-CoV-2 and IAV across major immune lineages. Hallmark inflammatory and interferon-stimulated genes are highlighted in orange and blue, respectively. e, Relative expression of IFN-α-encoding transcripts by each immune cell type in response to SARS-CoV-2 and IAV. Bar lengths indicate the mean number of IFN-α transcripts contributed by each cell type to the overall pool (cell type frequency × mean number of IFN-α transcripts per cell). Dot area is proportional to the mean level of IFN-α transcripts in each cell type (counts per million). f, Correlation of ISG activity scores between individuals, following exposure to SARS-CoV-2 and IAV. Each dot corresponds to a single individual (n = 222) and its color indicates the self-reported ancestry of the individual concerned (AFB: Central African; EUB: West European; ASH: East Asian).
Study findings
Genetics & Genomics eBook

Cellular proportions that varied due to environmental exposures were the primary drivers of population differences in SARS-CoV-2 immune responses. The higher proportions of memory cells detected in lymphoid lineages of Africans and their relationship with persistent cytomegalovirus (CMV) infection(s) suggested that population-level differences in cellular activation states might be driven primarily by lifelong pathogen exposure.
Socio-environmental factors were also found to covary with an individual's genetic ancestry, which, in turn, might lead to an overestimation of the effects on immune responses to SARS-CoV-2, which is a phenotypic variation.
Common genetic alleles also contribute to the observed variability of immune responses to viral challenges. However, their effects tend to be limited to a subset of genes displaying strong population differentiation.
For example, the rs1142888-G variant is found at a higher frequency in Europeans than Africans due to a selection event that happened between 21,900 and 35,600 years ago. This variant accounts for more than 2.8-fold higher levels of guanylate binding protein 7 (GBP7) expression that facilitates IAV replication by suppressing innate immunity.
GBP7 also regulates IFN-γ-induced oxidative host defense conferring resistance to intracellular bacteria, such as Listeria monocytogenes and Mycobacterium tuberculosis, thereby providing a feasible mechanism for positive selection at this gene locus.
Viral evolution changed the genetic basis of infectious diseases over time. Thus, limited overlap was observed between the alleles selected during this period in East Asia and the reported genetic variants underlying COVID-19 risk.
However, the researchers found traces of a selection event targeting SARS-CoV-2-specific reQTLs in East Asian ancestors that coincided with the proposed timing of an ancient epidemic about 25,000 years ago that affected the evolution of coronavirus-host interacting proteins.
Delineating the genetic architecture of immune response variations across several cell types provided mechanistic insights into the effect of COVID-19-associated alleles. For example, the efficiency of IFN signaling was confirmed to be essential for favorable clinical outcomes of SARS-CoV-2 infection.
A Neanderthal-introgressed eQTL at the mucin20 (MUC20) gene locus was found to increase its expression in SARS-CoV-2-stimulated CD4+ T-cells and decreased COVID-19 susceptibility. Perhaps the Neanderthal haplotype conferred higher resistance to viral infections through a similar effect, as mucins form a barrier against infection in the nasal epithelium.
Conclusions
Overall, the study findings highlight the significance of using sc-RNA-seq approaches to capture the diversity of the human immune response to RNA viruses, especially SARS-CoV-2. These observations provide new insights into the environmental, genetic, and evolutionary drivers of immune response variations across populations with genetic variations.
*Important notice
bioRxiv publishes preliminary scientific reports that are not peer-reviewed and, therefore, should not be regarded as conclusive, guide clinical practice/health-related behavior, or treated as established information.
- Aquino, Y., Bisiaux, A., Li, Z., et al. (2022). Environmental and genetic drivers of population differences in SARS-CoV-2 immune responses. bioRxiv. doi:10.1101/2022.11.22.517073. https://www.biorxiv.org/content/10.1101/2022.11.22.517073v1.
Posted in: Child Health News | Men's Health News | Genomics | Medical Science News | Medical Research News | Women's Health News | Disease/Infection News
Tags: Antibodies, Bacteria, Blood, CD4, Cell, Cell Migration, Coronavirus, Coronavirus Disease COVID-19, covid-19, Cytomegalovirus, Evolution, Frequency, Gene, Genes, Genetic, Genetics, Genome, Immune Response, immunity, in vitro, in vivo, Infectious Diseases, Influenza, Interferon, Interferons, Intracellular, Listeria, Locus, Neanderthal, Nucleotide, Pathogen, Protein, Respiratory, Ribonucleic Acid, RNA, SARS, SARS-CoV-2, Severe Acute Respiratory, Severe Acute Respiratory Syndrome, Single Nucleotide Polymorphisms, Syndrome, Tuberculosis, Virus

Written by
Neha Mathur
Neha is a digital marketing professional based in Gurugram, India. She has a Master’s degree from the University of Rajasthan with a specialization in Biotechnology in 2008. She has experience in pre-clinical research as part of her research project in The Department of Toxicology at the prestigious Central Drug Research Institute (CDRI), Lucknow, India. She also holds a certification in C++ programming.
Source: Read Full Article