
The evolution of a tumor
The results of evolution are often awe-inspiring—from the long neck of the giraffe to the majestic colors of a peacock—but evolution does not always create structures of function and beauty.
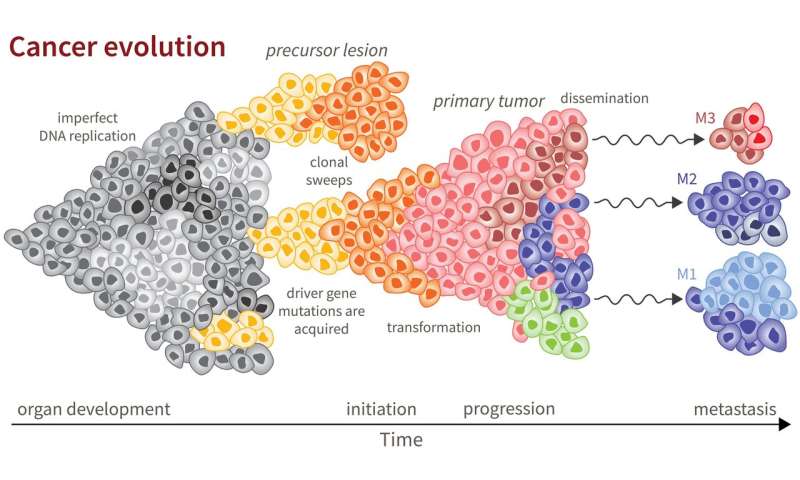
In the case of cancer, the growth of a population of malignant cells from a single cell reflects a process of evolution too, but with much more harrowing results.
Researchers like Johannes Reiter, Ph.D., of Stanford University’s Translational Cancer Evolution Laboratory, are examining the path of cancer from a single sell to many metastatic tumors. By using this perspective and simple mathematical models, Reiter interrogates the current practices in cancer treatment. He spoke at Duke’s mathematical biology seminar on Jan. 17.
The evolutionary process of cancer begins with a single cell. At each division, a cell acquires a few mutations to its genetic code, most of which are inconsequential. However, if the mutations occur in certain genes called driver genes, the cell lineage can follow a different path of rapid growth. If these mutations can survive, cells continue to divide at a rate faster than normal, and the result is a tumor.
With each additional division, the cell continues to acquire mutations. The result is that a single tumor can consist of a variety of unique cell populations; this diversity is called intratumoral heterogeneity (ITH). As tumors metastasize, or spread to other locations throughout the body, the possibility for diversity grows.
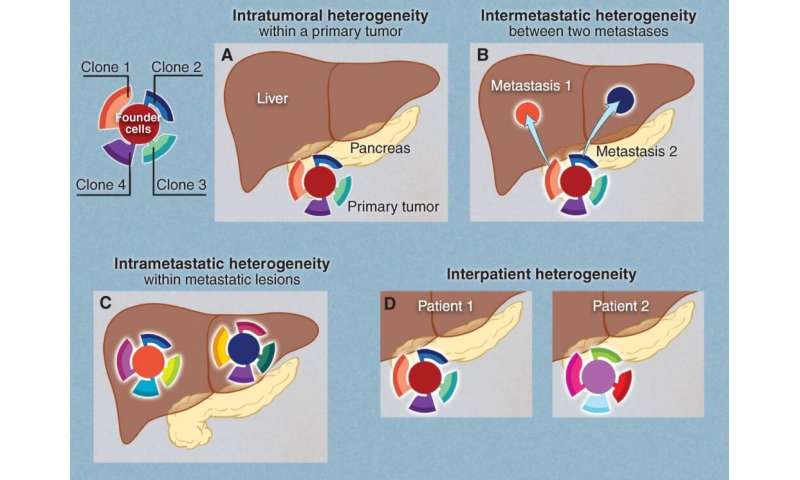
Reiter describes three flavors of ITH. Intra-primary heterogeneity describes the diversity of cell types within the initial tumor. Intra–metastatic heterogeneity describes the diversity of cell types within a single metastasis. Finally, inter-metastatic heterogeneity describes diversity between metastases from the same primary tumor.
For Reiter, inter-metastatic heterogeneity presents a particularly compelling problem. If treatment plans are made based on biopsy of the primary tumor but the metastases differ from each other and from the primary tumor, the efficacy of treatment will be greatly limited.
With this in mind, Reiter developed a mathematical model to predict whether a cell sample collected by biopsy of just the primary tumor would provide adequate information for treatment.
Using genetic sequence data from patients who had at least two untreated metastases and a primary tumor, Reiter found that metastases and primary tumors overwhelmingly share a single driver gene. Reiter said this confirmed that a biopsy of the primary tumor should be sufficient to plan targeted therapies, because the risk of missing driver genes that are functional in the metastases proved to be negligible.
Source: Read Full Article

